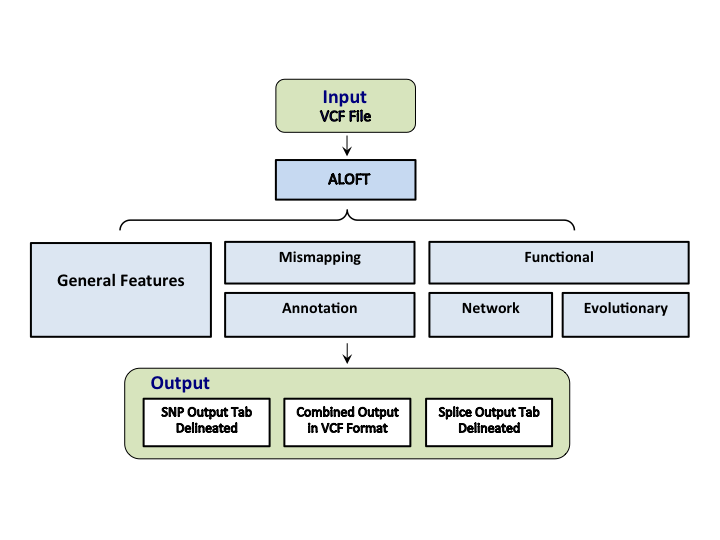
Annotation of Loss-of-Function Transcripts
ALoFT provides extensive annotations to putative loss-of-function variants (LoF) in protein-coding genes including functional, evolutionary and network features. Further, ALoFT can predict the impact of premature stop variants and classify them as dominant disease-causing, recessive disease-causing and benign variants.
Download ALoFT 1.0 Download ALoFT data (9.4GB)
Pre-calculated ALoFT scores (whole exome hg19)
Pre-calculated ALoFT scores (whole exome hg38)
Installation
Download ALoFT source code and data files then unzip both packages.
cd aloft/aloft-annotate python install.py
Add the following line to your .bashrc file, as explained at the end of the install.
export VAT_CONFIG_FILE=path_to_home_directory/.vatrc
Source your .bashrc file.
source .bashrc
To read the usage or to run ALoFT*:
./aloft --help ./aloft --vcf=/path/to/file --data=path/to/data/dir --output=path/to/dir [--option4=arg1]... ./aloft --vcf=/path/to/file --data=path/to/data/data_aloft_annotate --output=path/to/dir [--option4=arg1]...
*Note that the VCF file must contain a header according to VCF specification. The VCF file must also contain at least 1 LoF variant in order to successfully run to completion.
To install and run the prediction module of ALoFT (R must be previously installed):
cd aloft/aloft-predict
R
> install.packages(c("randomForest"))
perl aloft_predict.pl -i input -o output -d data_directory
perl aloft_predict.pl -i input -o output -d /path/to/data/data_aloft_predict
*Note that ALoFT predictions depend on features related to population allele frequency. In situations where more than one allele may cause a loss of function mutation at the same location, the reported ALoFT score will depend on the population frequency of the submitted allele.
See the documentation for more information.
Reference
Suganthi Balasubramanian, Yao Fu, Mayur Pawashe, Patrick McGillivray, Mike Jin, Jeremy Liu, Konrad J. Karczewski, Daniel G. MacArthur, and Mark Gerstein. "Using ALoFT to determine the impact of putative loss-of-function variants in protein-coding genes." Nature Communications 8 (2017).